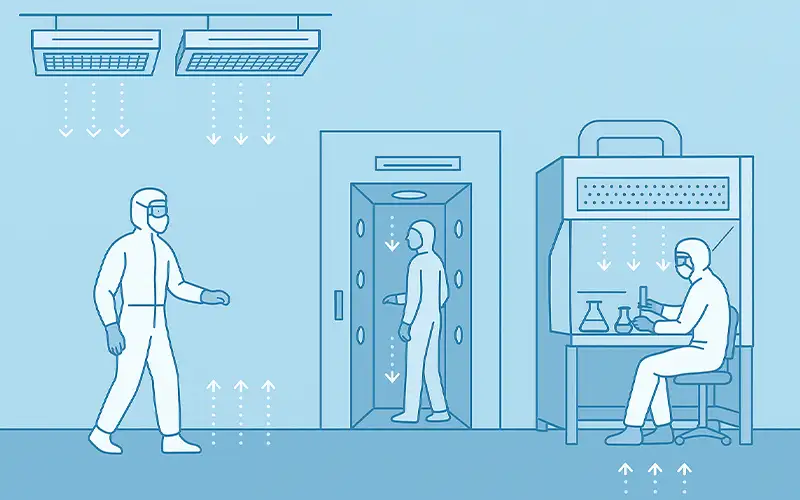
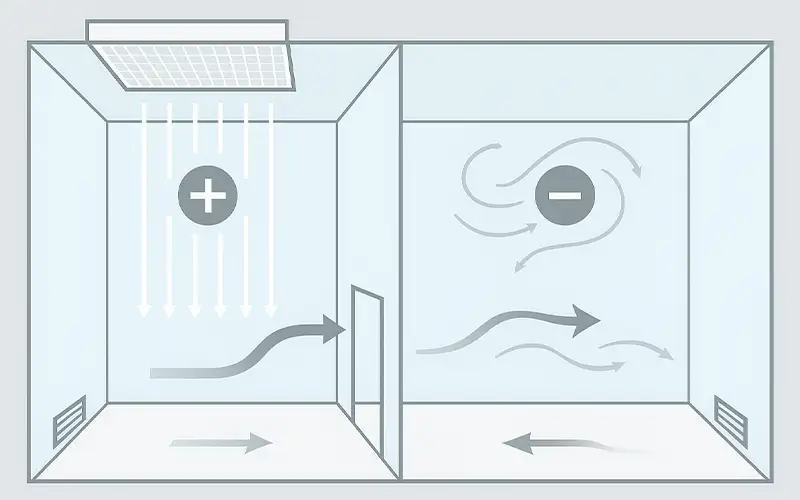
Prodotti per la Filtrazione dell'Aria
Esplorate la nostra gamma completa di soluzioni di filtrazione dell'aria di alta qualità, progettate per un'ampia varietà di settori industriali.
Dai filtri per HVAC alle applicazioni HEPA e industriali, i nostri filtri per l'aria offrono prestazioni superiori, efficienza energetica e un'affidabilità durevole.
Trovate il filtro perfetto per soddisfare le vostre esigenze.